
By Josh Wagner, Senior Quality Control Manager, Sterling Pharma Solutions
The author argues that a robust analytical approach is critical to not only determine when genotoxic impurities form, but also what their fate and tolerances may be.
Genotoxic impurities have become a major focus for pharmaceutical organisations and regulatory agencies alike. By definition, they are reactive with genetic material and may be carcinogenic or cause mutations when present in high enough concentrations. As a result, such impurities must be identified and eliminated to ensure they do not end up in final drug products where they could be a danger to patient safety through inadvertent exposure.
A robust analytical approach is critical to not only determine when these impurities form, but also what their fate and tolerances may be. They can come from a variety of sources: starting materials; catalysts or reagents used in manufacturing; reaction by-products or synthetic intermediates; or the result of degradation of a finished product. As a synthetic process evolves through early toxicological studies and clinical phases, so must the understanding of where these impurities have the potential to form. M7 guidelines released by the International Council for Harmonisation (ICH) in 2017 (1) give specific guidance on the assessment and control of mutagenic impurities from in silico assessment through in vivo testing, and set a high expectation on the burden of proof required.
When genotoxic impurities are identified in drug products once they have reached the market, it can cause widespread concern and a real risk to the health of millions of patients. In 2018, the Federal Drug Administration (FDA) and European Medicines Agency (EMA) recalled drug products that contained valsartan, commonly prescribed to treat high blood pressure, following the identification of the carcinogen N-nitrosodimethylamine (NDMA) in a number of batches. This triggered wider industry concern about potential NDMA contamination in other drugs, including ranitidine and metformin, and led to the EMA providing guidance to marketing authorisation holders. This included how to avoid the presence of nitrosamine impurities in human medicines and asking for risk assessments for all human medicines to determine the potential for nitrosamines to be present. It also required the testing of products identified to be at risk (2).
Besides the risk to human health, identifying these impurities late in development can delay clinical progression and regulatory approval. After commercial launch, they can result in significant remedial efforts, including potential product hold and recall. Consequently, accurate impurity assessments throughout development and robust control measures are essential to mitigate genotoxic impurities during manufacturing.
Categorisation
Impurities are categorised into five classes based on an assessment of the risk they pose, as shown in the table. Class 1 represents the greatest risk, based on established mutagenic and carcinogenic data. These impurities should be completely eliminated wherever possible, or if they cannot be fully removed, should be significantly limited to below the threshold of toxicological concern (TTC), which is defined by regulatory bodies. Class 5 impurities hold the lowest risk potential, with nothing to indicate genotoxicity, and should be treated as typical impurities that can arise in drug products.

Classification of Genotoxic Impurities (3).
Regulatory guidance
Unlike other impurities, such as residual metals in drug products, regulatory guidance regarding genotoxic impurities is relatively new. The EMA and FDA released guidelines in 2006 and 2008, respectively, which were preceded by a Pharmaceutical Research and Manufacturers of America (PhRMA) paper (4) that discusses controlling impurities and classifies them into the five categories shown in the table above. In general, the guidance focused on limiting genotoxic impurities below the TTC, which the FDA and EMA have both set as 1.5 μg / person / day (5) rather than eliminating them entirely.
Today, the main regulatory focus is the ICH M7 guidelines, which resolve some inconsistencies in the previous guidance and provide detailed recommendations for testing and controlling impurities to minimise the level of carcinogenic risk in finished products.
Identification of genotoxic impurities
Potential and known genotoxic impurities are treated differently. Known impurities should always be tested for at an appropriate control point, with a defined specification. Potential genotoxic impurities (PGIs) are theoretical, either in predictive toxicity or actual presence, and their inclusion depends on the point in synthesis at which they are likely to occur and/or purge.
As previously noted, there are many potential sources of these impurities, so identifying and controlling PGIs is particularly challenging. Prediction of how and when they may arise requires a deep synthetic knowledge, coupled with a variety of means of evaluating their potential for genotoxicity. As many of these can be avoided through route selection and controlled through process design, it is important to consider their presence throughout a product’s lifecycle. A typical assessment will begin with a review of reagents, intermediates, known impurities and potential synthetic impurities that can identify all potential chemical species that may be present in the API. This list can then be assessed against known genotoxins and evaluated with quantitative structural activity relationship (QSAR) software programs for structural alerts.
There is a range of chemical structural moieties that should prompt further genotoxic testing, with some of the most common being aromatic nitro groups, aromatic amines, Michael acceptors and alkylating agents.
If an impurity triggers a structural alert, an Ames test is necessary to further assess mutagenicity. This test uses a Salmonella strain to determine whether induced reversion mutations form in the presence of the mutagen. This is a highly sensitive technique, detecting mutation rates as low as 10-7/cell. The level of mutagenicity compares the rate of induced mutation compared to the spontaneous mutation as the standard. Mutations can occur at extremely low concentrations of a mutagen and conservatively protective rates of incident have been used to establish acceptable exposure levels.
Because low quantitation limits must be applied to ensure established acceptable daily exposures (ADEs) are not exceeded, developing appropriate and robust analytical methods is essential. Specifications are based on the product’s maximum daily dose and may be introduced at an appropriately identified point of control. While early in development, these factors are likely to change as the final dose, dosage form and process evolves. However, performing the assessment and establishing scientifically-sound methods from the outset can ensure these impurities are considered in the process design and fate and tolerance, and can be established throughout the process. This reduces the risk of significant changes late in development, potentially impacting validity of registrational stability or the timing to file for commercial approval.
Analytical approaches
These impurities can have a variety of structures and variants, but often, by their very nature of reacting with DNA, are chemically reactive. This can make finding analytical methods that have good recovery and an appropriate quantification limit (QL) challenging. This means that any analytical approach must be highly tailored.
The first requirement for any analytical test must be that it can identify potential impurities at concentrations well below the acceptable daily exposure level. While it requires low limits of detection, a suitably wide quantitation range is important too as limits may evolve over time, even post commercialisation. It can also give the opportunity to support tolerance arguments that eliminate the need for routine testing or further assessment of PGIs. Typically, specifications are in the parts per million range.
Secondly, while specificity is always needed, it is particularly pertinent for genotoxic impurities. As limits are low, any interfering impurity will almost certainly result in a false failure. Clean chromatographic baselines and high levels of baseline resolution are a focus in established high quality methods.
A variety of analytical techniques can be deployed such as high-performance liquid chromatography (HPLC) with ultraviolet-visible (UV-Vis) detection, as well as high-sensitivity detectors such as mass spectrometers. Gas chromatography with flame ionisation detectors (FID) or mass spectrometers are also common approaches. Sometimes, a combination of analytical methodologies may be needed to quantify all known and potential impurities.
Guidance is clear on the need to control these impurities throughout both process and testing. However, each process, and therefore control strategy, is unique and must be defined by the drug sponsor. To provide a clearer image of what testing approach to leverage at which stage in development, a flowchart developed by Reddy et. Al (4) explains a rationale for analytical methodology based on when PGIs surface. For example, if the impurity is introduced in the final step, it is critical to test the API itself for the presence of impurities and impose a limit on impurity concentrations.
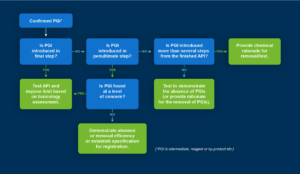
The analytical methodology for PGIs differs based on when the PGI is introduced.
If an impurity is introduced in a penultimate step and found at a level of concern, the same testing approach should be utilised as if introduced in the final step. If it is not present at a level of concern, then it is vital to demonstrate its absence with robust supporting data and analysis. If it surfaces earlier in the synthesis, then a rationale for its removal must be shown. If no PGIs are identified, then it is important to generate supporting data that proves the absence of genotoxic impurities.
As the industry and regulatory bodies evolve, their philosophies and approaches to drug development, higher risk aversion and more patient-protective strategies will continue to expand the expectations for process characterisation and control. Taking a robust analytical approach from the start will help to mitigate project challenges in the long run and deliver the evidence and analyses needed to gain regulatory approval.
- Https://www.ema.europa.eu/en/ich-m7-assessment-control-dna-reactive-mutagenic-impurities-pharmaceuticals-limit-potential
- https://www.ema.europa.eu/en/human-regulatory/post-authorisation/referral-procedures/nitrosamine-impurities
3, Gosar, A.; Sayyed, H.; Shaikh, T. Genotoxic Impurities and Its Risk Assessment in Drug Compounds. Drug Des Int Prop Int J [Online] Oct. 23, 2018, 227-232.
- Reddy, A. et. al. Identification, control strategies, and analytical approaches for the determination of potential genotoxic impurities in pharmaceuticals: A comprehensive review. J. Sep. Sci. [Online] 2015, 00, 1-16
- https://pubmed.ncbi.nlm.nih.gov/16412543/
