
By our Editorial Team
Nicola Travierso, President of Velit Bio, discusses the market dynamics of, and opportunities in the future for, biosimilars

Nicola Travierso, President of Velit Bio, discusses the market dynamics of, and opportunities in the future for, biosimilars in the US and Europe.
The current global biologics market is worth $127 billion ($60 billion in the US alone, and $35 billion in Europe), and it is growing at a rate of around 9% a year.
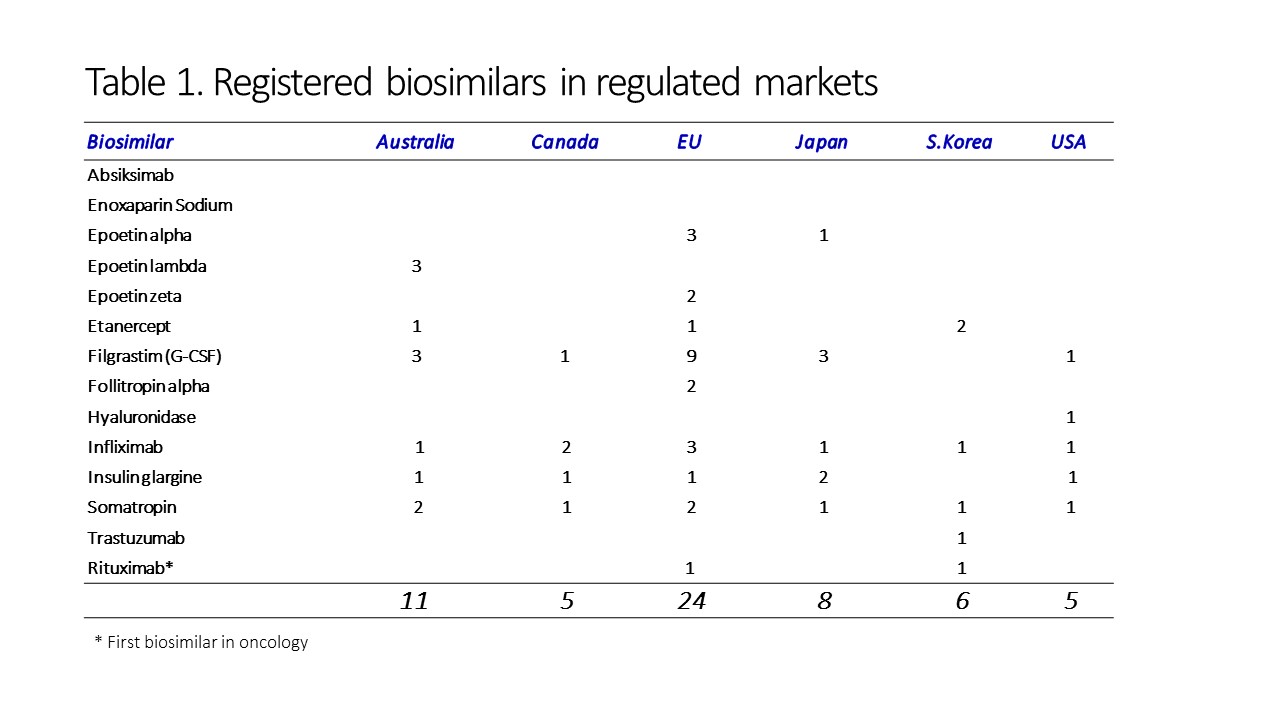
At the time of writing this article, 14 different biosimilars have been approved and registered in regulated markets across the world (Table 1). However, it is interesting to note that while the US shows the greater expenditure on biosimilars, this is based on a market of just five FDA-approved products (Celltrion’s infliximab-dyyb, Sandoz’s filigrastin-sndz and etanercept-szzs, Amgen’s adalimumab-atto, and Samsung Bioepsis’ infliximab-abda). Europe, on the other hand, has 24 approved individual products already, and the number is growing.
As with any pharmaceutical product, the development of a biosimilar needs to begin with non-clinical studies. For biosimilars, the purpose of non-clinical phase research – primarily pharmacodynamic and toxicological studies – is to detect differences between the biosimilar and the reference biologic product. Assuming that the biosimilar is shown to be ‘similar enough’ to the reference biological, R&D proceeds to the clinical phase, with the possibility to extrapolate findings from non-clinical, pharmacokinetic and pharmacodynamic studies, which allow for the conclusion of similar efficacy and safety.
“These guidelines are implemented in the US, Europe and Japan,” explains Mr Travierso. “For each biosimilar under development the relevant regulatory agencies have an open discussion on whether more than one clinical trial is required, but typically no further clinical data are required if the pharmacokinetic and pharmacodynamic data allow for conclusions of efficacy and safety – and assuming the impurity profile and the nature of the product’s excipients do not give rise for concern.”
The lower costs associated with the development and approval of biosimilars is reflected in the lower prices for these products.
“Eating into a significant proportion of the biologic market in Europe is the practice of ‘switching’ biosimilars for more costly reference products,” says Dr Travierso.
